Article Text

Abstract
Alexander disease is a rare, genetic and ultimately fatal neurological disorder that arises from pathogenic variants in the glial fibrillary acidic protein (GFAP) gene. Its presenting symptoms often differ according to age at onset. Although Alexander disease typically presents in young children with seizures and developmental delays, its presentation in adults may include bulbar signs, ataxia and autonomic dysfunction. Because of the heterogeneous and non-specific symptoms associated with adult-onset Alexander disease, the diagnosis typically requires comprehensive clinical and neuroimaging evaluation as well as confirmatory genetic testing. Here, we present detailed case descriptions of patients who first presented with symptoms of Alexander disease as adults, with guidance on recognising distinctive clinical and radiological characteristics associated with the later-onset form. Timely recognition and referral of patients with Alexander disease will enable earlier interventions that may mitigate disease severity or slow disease progression if such interventions become available.
- NEURORADIOLOGY
- MRI
- MOVEMENT DISORDERS
- CLINICAL NEUROLOGY
- NEUROGENETICS
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Alexander disease (AxD) is a rare, progressive leukodystrophy caused by pathogenic variants in the glial fibrillary acidic protein (GFAP) gene. Its pathophysiology is characterised by excess production and accumulation of GFAP in astrocytes,1 leading to the formation of Rosenthal fibres, a characteristic histopathological feature first reported in a young patient by William S. Alexander in 1949.2 3 AxD may present with a range of symptoms and neuroimaging features, which generally fall into patterns according to age of onset.4–6 In infants and young children, it typically manifests with seizures and developmental delays,4 5 and MRI findings characteristically include frontally predominant white matter abnormalities.7 The clinical diagnosis of AxD in those first presenting as adults is often challenging because of the non-specific nature and heterogeneity of symptoms, and comprehensive evaluation including careful review of clinical presentation and neuroimaging findings is crucial for diagnosis.3 Although AxD is rare, neurologists may encounter many patients with conditions of unclear cause whose clinical presentation overlaps with that of AxD. The diagnosis in these patients is complex and may require significant amounts of time and multidisciplinary subspecialty evaluations.
Given the progressive and disabling nature of AxD, early and accurate diagnosis may become more critical as an ongoing clinical trial investigating a potential disease-modifying therapy for AxD could result in new opportunities for treatment. To increase awareness of the distinctive characteristics and neuroimaging patterns seen in adults with AxD, we reviewed the records of patients who first presented as adults with AxD symptoms, aiming to highlight the range of clinical characteristics, neuroimaging features and potential differential diagnoses in such cases. Here, we review and describe the spectrum of AxD presentations through detailed case descriptions and provide guidance on recognising clinical and radiologic pearls to aid in timely recognition and referral of patients with suspected AxD.
Clinical spectrum of AxD
AxD can present at any time throughout a person’s lifespan from birth to late adulthood,8 with adolescents and adults (≥12 years of age) accounting for up to 41% of reports.9 The clinical spectrum of AxD is phenotypically heterogeneous and may include symptoms of bulbar, motor, autonomic, gastrointestinal and cognitive dysfunction.2–4 6 In most patients with a later age at onset, the disease progresses more slowly over several years to decades; however, the disease course can be varied and unpredictable, and some patients with adult onset may show rapid progression and death within several years of diagnosis.3
In infants and young children, typical indicators of AxD include seizures, developmental delay and macrocephaly,4 5 although not all patients have these symptoms at clinical onset.10 Patients with AxD presenting first in adulthood are more likely to have bulbar signs, ataxia and autonomic dysfunction.2–4 Palatal myoclonus strongly suggests AxD in adults, although it is not always present.3 In juvenile and adolescent patients, symptoms may resemble those associated with either childhood-onset or adult-onset disease, or a combination of both.2 6 In some cases, patients diagnosed with AxD as adults have a history of milder cognitive or developmental symptoms (eg, poor school performance, clumsiness) during childhood or adolescence. Intractable vomiting can be a presenting symptom and can occur in isolation.5
Although many adult patients with AxD present with bulbospinal features (characterised by muscle weakness, hyper-reflexia and bulbar signs), the clinical presentation can vary significantly from patient to patient. Some patients have atypical (eg, neuropsychiatric) or non-specific symptoms at onset.11 The neuropsychiatric presentation of AxD can include depression, behavioural dysregulation, cognitive dysfunction and memory impairments, which may be misdiagnosed as a psychiatric disorder (eg, major depressive disorder, postpartum depression).11 Sometimes there are varied clinical presentations within the same family.12 Because of the heterogeneity in clinical presentation, patients may initially receive inaccurate diagnoses or have a history of misdiagnosed family members, highlighting the difficulty of diagnosing AxD based on clinical assessment alone3 13 and emphasising the importance of MR imaging in establishing a diagnosis.
Neuroimaging findings in adult-onset AxD
In patients with suspected AxD or those presenting with neurological symptoms of unclear cause, it is critical to identify MRI features characteristic of AxD to prompt genetic testing for pathogenic variants in GFAP or referral to a specialist to establish a definitive diagnosis (figure 1). Neuroimaging findings in older children, adolescents and adults often differ from the classic leukodystrophy pattern observed in younger children, which is typically characterised by frontally predominant T2 hyperintensities as well as basal ganglia T2 signal abnormalities and swelling (figure 2), and more likely includes signal intensity changes or atrophy of the brainstem, cervical spinal cord and/or cerebellar structures.2 3 14 Marked atrophy of the medulla oblongata and cervical spinal cord with sparing of the basis pontis (ie, ‘tadpole sign’) is a characteristic finding in patients with AxD, particularly in those presenting as adults.15 Other supportive radiologic signs in adults with AxD may include signal changes in the basal ganglia or thalami16 17 and/or the periventricular white matter.2 3 14 16 17 Affected structures may show contrast enhancement.2 3 14 16 Although uncommon, some adult patients may have confluent cerebral white matter abnormalities with a frontal predominance similar to those typically present in early onset AxD.2 15 There are also reports in the literature of a range of atypical MR imaging features in adult patients with AxD, including ventricular garlands,16–18 dilatation of the lateral ventricles2 and hydromyelia.19

Clinical pathway for diagnosis and early management for a patient with suspected AxD. AxD, Alexander disease; GFAP, gene encoding glial fibrillary acidic protein.

MR scan of the brain of a patient with infantile AxD. This patient with cerebral (or infantile) AxD has frontally predominant T2 hyperintensities of the white matter and basal ganglia, with additional swelling of the caudate bilaterally. AxD, Alexander disease.
Illustrative cases
Case 1: ataxic presentation with brainstem and spinal cord atrophy
A man in his early 40s presented with progressive worsening of balance, paraesthesia in both arms and substantial fatigue. Over the next 2 years, his mobility worsened, particularly because of spasticity on his right side. He could walk only half a mile before feeling exhausted, and he began using a cane. There were no bulbar features or sphincter problems and no suggestion of cognitive involvement. His medical history included pernicious anaemia and hypertension. His family history included reported multiple sclerosis in two close relatives.
On examination, he had a normal facial expression and full range of eye movements. There was no palatal tremor. His voluntary movements were slower, suggesting pyramidal weakness and spastic tone was mildly to moderately increased on the right side. Muscle strength was normal on the left side; however, there was mild weakness of right shoulder abduction and hip flexion. Sensation was normal throughout despite subjective complaints. He had diffuse hyper-reflexia, more pronounced on the right and an extensor right plantar response. His gait was mildly broad-based.
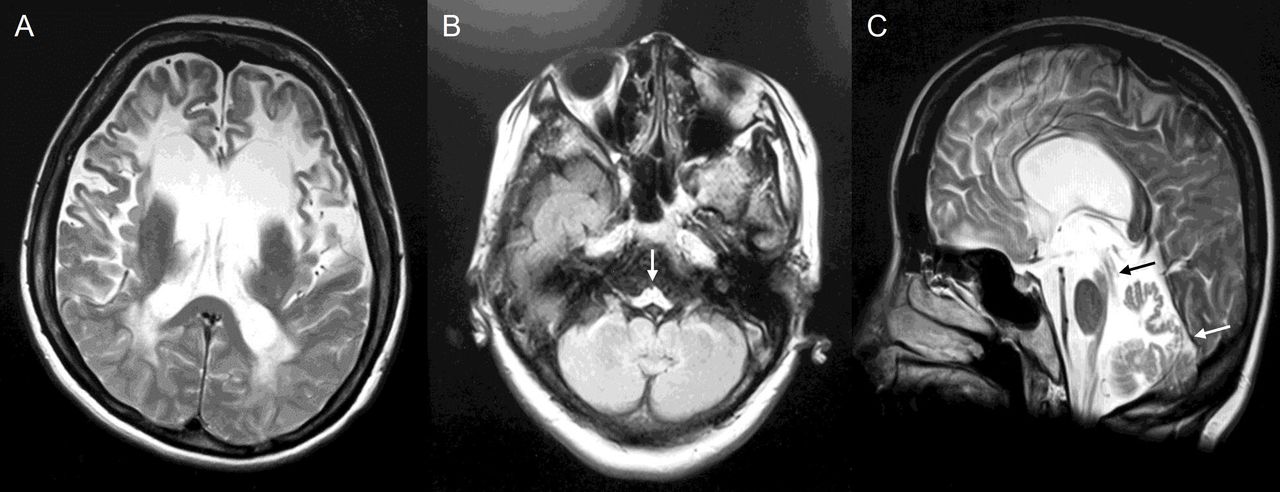
MR scan of the brain 2 years after symptom onset showed scattered T2-weighted hyperintensities, including in the pons and middle cerebellar peduncle, with marked volume loss in the brainstem and spinal cord, and some cerebellar atrophy (figure 3). Given the imaging findings, the treating physician requested genetic testing with a leukodystrophy panel and identified a c.1154C>G p.Ser385Cys GFAP variant.20

MR scan of the brain of case 1. Axial and sagittal FLAIR images showed only moderate periventricular abnormalities supratentorially (A). Infratentorially, (B, C) there were scattered T2 hyperintensities in the pons, medullary pyramids (arrow in B), dentate nuclei, and middle cerebellar peduncles (arrow in C), with marked loss of volume in the medulla and cervical cord. FLAIR, fluid-attenuated inversion recovery.
Clinical pearls
Patients may report parents or other family members who have balance or gait problems or even potentially erroneous diagnoses such as multiple sclerosis, underscoring the need to be suspicious of family history. Clinicians should be especially cautious about historic diagnoses in family members from an era before widespread genetic testing was available. The top clinical differential in this case was primary progressive multiple sclerosis.
Case 2: AxD mimicking spinocerebellar degeneration
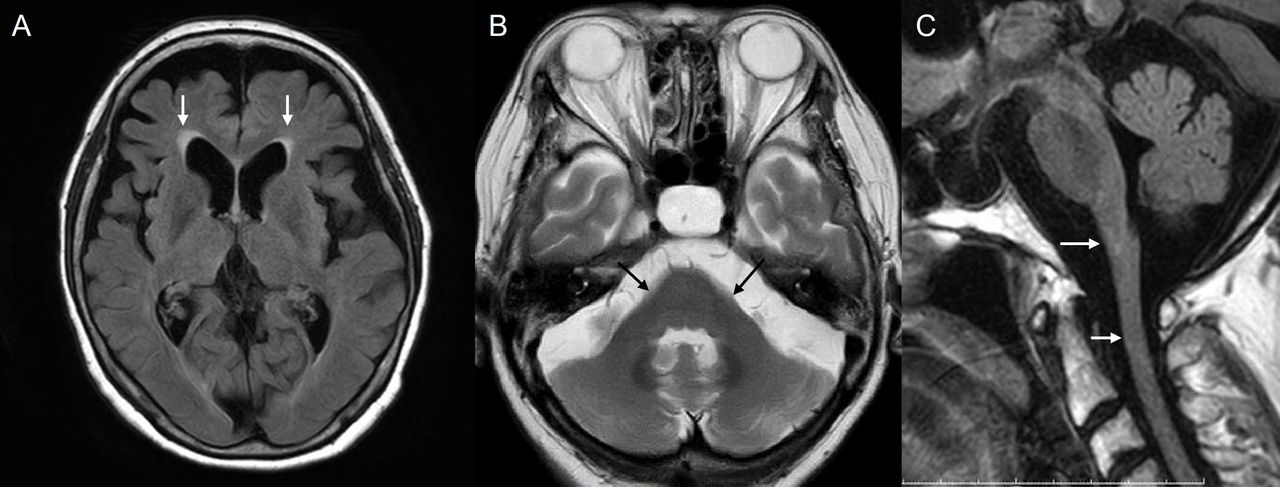
A woman in her early 50s was referred following abnormal findings on brain MRI performed during hospitalisation for respite care. She had previously presented in her late 30s with repeated falls, and 2 years later was diagnosed with spinocerebellar degeneration. 5 years after initial presentation, she became unable to walk independently and required a wheelchair. 7 years after initial presentation, she underwent a gastrostomy because of severe dysphagia. She had no family history of any neurological conditions. At the time of neurological assessment, she was confined to her bed, with orthostatic hypotension, bladder dysfunction, severe dysarthria with tongue atrophy, palatal myoclonus and spasticity of the limbs with positive extensor plantar responses. MR scan of the brain showed severe atrophy of the medulla oblongata, cerebellum and midbrain as well as T2 hyperintensity of the cerebral white matter (figure 4). GFAP gene analysis identified a heterozygous c.250 G>A p.Arg79His missense variant.20

MR scan of brain of case 2the, showing cerebral white matter lesions predominantly in the frontal lobe (A), and severe atrophy of the medulla oblongata (arrow in B), cerebellum (white arrow in C) and midbrain (black arrow in C) with preservation of the pons (C).
The history later obtained from the patient’s mother found that the patient had experienced febrile convulsions from infancy to around 5 years of age and had undergone surgery for scoliosis as an adolescent. Furthermore, her mother had been concerned that the patient might have had an unsteady gait and intellectual disability when of school age.
Clinical pearls
Childhood-onset slowly progressive cerebellar ataxia in patients with AxD can be misdiagnosed as spinocerebellar degeneration, including recessive spinocerebellar ataxia. A history of childhood febrile seizures may be a clue to suspect that the patient might have AxD.
Case 3: AxD with syncope as an initial symptom
A woman in her early 70s was referred with episodic syncope and progressive gait disturbance. She had lost consciousness on standing 3–4 times per year for 2 years before the initial evaluation. She had been hospitalised for a head injury because of syncope 2 years before the referral, at which point an MR scan of the brain had identified cerebellar atrophy. One year later, she was aware of gait disturbance and speech disturbance. 2 months before the evaluation, she could walk only with support. She had no family history of any neurological conditions. Neurological examination showed saccadic eye movements, dysarthria and limb ataxia as well as spasticity of the lower extremities with positive extensor plantar responses, orthostatic hypotension and bladder dysfunction. MR scan of the brain 3 years after symptom onset showed atrophy and T2/fluid-attenuated inversion recovery (FLAIR) signal abnormality of the medulla oblongata and upper cervical cord, as well as atrophy of the cerebellum and middle cerebellar peduncle (figure 5). GFAP gene analysis identified a homozygous c.740 C>T pSer247Phe missense variant.20

MR scan of brain of case 3 showing changes consistent with AxD including (A) focal symmetric juxtaventricular white matter lesions capping the anterior horn of the lateral ventricles (arrows in A), (B) atrophy of the cerebellum and middle cerebellar peduncles (arrows in B) and (C) mild atrophy of the medulla oblongata and upper cervical cord (arrows in C) with preservation of the pons. AxD, Alexander disease.
Clinical pearls
Adults with AxD may present with autonomic dysfunction, such as repeated fainting, as the initial symptom. This may be diagnosed as autonomic failure, multiple system atrophy, spinocerebellar degeneration or isolated syncope.
Case 4: spinobulbar presentation with periventricular white matter abnormalities
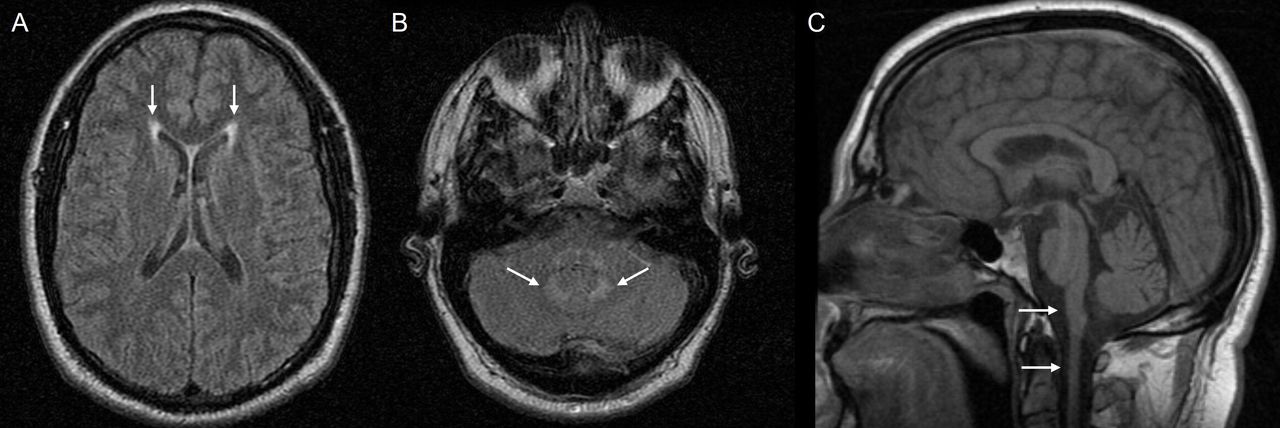
A man in his early 50s sustained a fall with loss of consciousness. When he awoke, he had unintelligible speech, right-sided weakness and drooling, which quickly improved. He was taken to a local hospital where a vascular cause was suspected. He had a history of sleep apnoea requiring continuous positive airway pressure. An MR scan of the brain without contrast showed periventricular white matter T2/FLAIR hyperintensities, attributed to mild chronic small vessel ischaemic disease. About 4 months later, he developed swallowing difficulties, voice changes and questionable aspiration. He was evaluated by otolaryngology and was diagnosed with spastic dysphonia and palatal myoclonus. Swallow study confirmed aspiration. He was then referred to neurology and was noted to have an enhanced jaw jerk as well as a palatal tremor, dysmetric saccades, dysdiadochokinesia and dysmetria. He also had subtle, symmetric, upper motor neurone distribution weakness, brisk myotatic stretch reflexes and bilateral Hoffmann signs, all in the upper limbs. He had normal patellar but brisk Achilles reflexes. He was suspected to have possible adult-onset AxD. An MR brain scan, which was not available for review, was notable for mild volume loss and hyperintensities in the midbrain and medulla oblongata as well as scattered punctate T2/FLAIR hyperintense foci in the periventricular and subcortical white matter (repeat imaging at 6 months is shown in figure 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MR scan of brain of case 4. Brain imaging 6 months after the initial presentation (fall with loss of consciousness and transient neurological symptoms) revealed subtle changes consistent with AxD including (A) bilateral, symmetric periventricular hyperintensities (arrows in A). (B) pontocerebellar hyperintensities (arrows in B) and (C) medullary/upper cervical cord atrophy (arrows in C). AxD, Alexander disease.
Clinical pearls
AxD may occasionally present with falls, which can be a symptom of autonomic dysfunction. Palatal myoclonus strongly suggests but is not pathognomonic for AxD, and its presence warrants additional testing. Imaging abnormalities can be subtle; clinicians should pay careful attention to the medulla, including in both the axial and sagittal planes to assess for medullary atrophy.
When to suspect adult-onset AxD
Adults with AxD often experience delays in diagnosis or misdiagnosis because of its sometimes subtle and variable presentation. Table 1 lists the typical clinical findings of adult-onset AxD. Although the clinical presentation can vary, adults with AxD often share a common clinical pattern characterised by ataxia, bulbar signs and spasticity.2 Motor symptoms or gait abnormalities are frequent but may be subtle. Many adults are described as ‘clumsy’ and may have occasional falls, which can be a symptom of dysautonomia or mild ataxia. Unexplained syncope or orthostatic hypotension should prompt brain imaging as well as autonomic testing. Important clues may be hidden in a patient’s history; for example, recurrent urinary tract infections may hint at upper motor neurone bladder dysfunction or sleep apnoea may potentially be linked to brainstem involvement. Not all patients with adult-onset AxD have frontally predominant white matter hyperintensities. Instead, supratentorially, there may be subtle periventricular cerebral white matter changes, as did several of the cases presented. The characteristic imaging findings in the brainstem, such as T2 signal change and atrophy of the medulla and cervical spinal cord, should trigger suspicion of the diagnosis; however, the MR brain scan may appear normal in some patients with AxD. AxD should be considered even before neuroimaging in patients with certain red flag symptoms, such as palatal myoclonus or a suggestive family history.
Common symptoms and localisation of AxD in adults
Differential diagnosis in adults with suspected AxD
The clinical context and neuroimaging findings will determine the differential diagnosis in a patient with suspected AxD. If there is prominent white matter signal change, then alternative leukodystrophies should be considered, and a leukodystrophy panel would be appropriate.21 Multiple sclerosis is the most common acquired disorder that may have overlapping features, including hemiparesis, dysarthria and ataxia, although it can be distinguished from AxD on imaging by the presence of asymmetrical findings, thoracic cord lesions or small optic nerve lesions (table 2).22 23 Neuromyelitis optica spectrum disorder could also be considered, especially given the overlapping presentation of the area postrema syndrome24 25 and the intractable vomiting seen in some patients with AxD.26 In general, AxD is associated with negative inflammatory biomarkers (eg, absence of cerebrospinal fluid oligoclonal bands), which may help to distinguish it from inflammatory and immune-related disorders.27 28 There are also many neurodegenerative disorders with overlapping clinical features, in particular ataxias like the spinocerebellar ataxias, Parkinsonian syndromes, multiple system atrophy and dementias.29 30 Cognitive and bulbar presentations, as well as brainstem and cerebellar signal change and brainstem atrophy, often occur in adult-onset polyglucosan disease.31
Differential diagnosis of AxD in adults
Understanding genetic testing
Genetic testing should be performed when possible to confirm the diagnosis of AxD in patients with suggestive clinical and neuroimaging features (ie, brainstem and spinal cord abnormalities).32 If there is a known family history, then single-gene testing may be appropriate; however, for most patients a multigene panel (eg, leukodystrophy panel including GFAP as well as other genes of interest) will be more efficient and comprehensive, and therefore, more likely to result in a confirmed diagnosis. In regions such as England where whole-genome sequencing is now routine for genetic testing, GFAP is included in a diverse array of targeted genetic panels including inherited white matter disorders, childhood or adult-onset ataxia, movement disorders and dementia among others, which reflects the heterogeneous presentations of AxD. However, this is not the case in other regions. Clinicians in regions such as the USA must, therefore, remain aware of the limitations of some genetic tests, as GFAP is included in some, but not all, commercial ataxia panels. While GFAP is included on most leukodystrophy panels, clinicians may not consider a leukodystrophy if brain imaging shows no significant white matter involvement, such as the cases described here presenting with ataxia. Thus, initial negative testing using a panel approach in a patient with ataxia should prompt additional, more comprehensive testing, such as a larger panel or whole-exome sequencing or whole-genome sequencing.21 Because of the complex and challenging process involved in diagnosing AxD, collaboration when possible among neurologists, neuroradiologists and medical geneticists familiar with AxD is likely to improve diagnostic accuracy.
Conclusions
The diagnosis of AxD in adults may be complex and challenging. Previous diagnoses in the cases presented here included spinocerebellar degeneration and spastic dysphonia. Although some patients had a family history of AxD, others did not or had family members who had also been misdiagnosed with multiple sclerosis. Some patients, as in case 2, may also have a history of subtle or mild symptoms during childhood that were unrecognised as potential AxD until worsening in adolescence or adulthood. AxD may also present with rapid clinical onset or progression and/or stroke-like symptoms, particularly in association with falls or mild head trauma.33 These examples highlight the importance of observing best practices in recognising key clinical features (eg, components of history and neurological examination) and MR brain scan features to identify patients suspected to have AxD. Awareness of distinctive clinical and radiologic characteristics associated with AxD among neurologists and neuroradiologists will enable earlier implementation of interventions that may reduce disease severity and/or modify disease progression if such interventions become available.
Key points
The presenting symptoms of Alexander disease often differ with its age at onset; although typically in young children it presents with seizures and developmental delays, its presentation in adults may include bulbar signs, ataxia and autonomic dysfunction.
Clinicians should consider Alexander disease in patients with certain ‘red flag’ features, such as palatal myoclonus; also, characteristic imaging findings in the brainstem, such as T2 signal change and/or atrophy of the medulla and cervical spinal cord should trigger suspicion of the diagnosis (table 1).
Some patients with Alexander disease have subtle presenting symptoms, such as falls or general clumsiness, or a history of mild symptoms during childhood not recognised as potential Alexander disease until worsening in adolescence or adulthood.
The clinical context and neuroimaging findings determine the differential diagnosis; multiple sclerosis is the most common acquired disorder with overlapping features, although imaging distinguishes it from Alexander disease, with asymmetrical findings, lesions involving the thoracic cord or small optic nerve lesions (table 2).
Clinicians should remain aware of the limitations of genetic testing, which may not include GFAP; initial negative panel testing in a patient with ataxia should prompt additional, more comprehensive testing (eg, whole-exome or whole-genome sequencing).
Further reading
Pareyson D, Fancellu R, Mariotti C, et al. Adult-onset Alexander disease: a series of eleven unrelated cases with review of the literature. Brain 2008;131:2321-31.
Muthusamy K, Sivadasan A, Dixon L, et al. Adult-onset leukodystrophies: a practical guide, recent treatment updates, and future directions. Front Neurol 2023;14:1219324
Lynch DS, Wade C, Paiva ARB, et al. Practical approach to the diagnosis of adult-onset leukodystrophies: an updated guide in the genomic era. J Neurol Neurosurg Psychiatry 2019;90:543-54.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Acknowledgments
Manuscript preparation and editorial support were provided by Dana Lengel, PhD, of Red Nucleus and funded by Ionis.
References
Footnotes
X @alisecarlsonmd
Contributors All authors participated in drafting, revising or critically reviewing the manuscript. All authors reviewed and approved the final draft for submission. DSL is the guarantor.
Funding This literature review was funded by Ionis Pharmaceuticals.
Competing interests DSL is a consultant and scientific advisor to Eli Lilly and Vigil Neuroscience. AKC has received research support from Biogen and Genentech; has served on scientific advisory boards for Bristol Myers Squibb and Sanofi; and has served as a consultant for Icometrix, Ionis, Novartis and Vigil Neuro. FB is a steering committee or data safety monitoring board member for ATRI/ACTS, Biogen, Merck and Prothena; serves as a consultant for Celltrion, Combinostics, IXICO, Janssen, Merck, Rewind Therapeutics and Roche; he also has research agreements with Biogen, GE Healthcare, Merck and Roche; and is a cofounder and shareholder of Queen Square Analytics Ltd. AC and MRE are employees of and hold stock in Ionis. ATW has received grant funding from the NIH (U54NS115052); she has also received research funding for investigator-initiated research from Elise’s Corner, Grayson’s Ladder, Ionis and the United Leukodystrophy Foundation; she has received clinical trial support from Ionis and (unrelated to this work) Novartis, Passage Bio, Pfizer, Roche/Genentech and Sarepta. She has served as a non-remunerated scientific advisor for Elise’s Corner and The Calliope Joy Foundation; receives honoraria from MedLink Neurology and UpToDate and received personal compensation for serving on a data safety monitoring board from SwanBio. All other authors report no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed by Marios Hadjivassiliou, Sheffield, UK.